Background
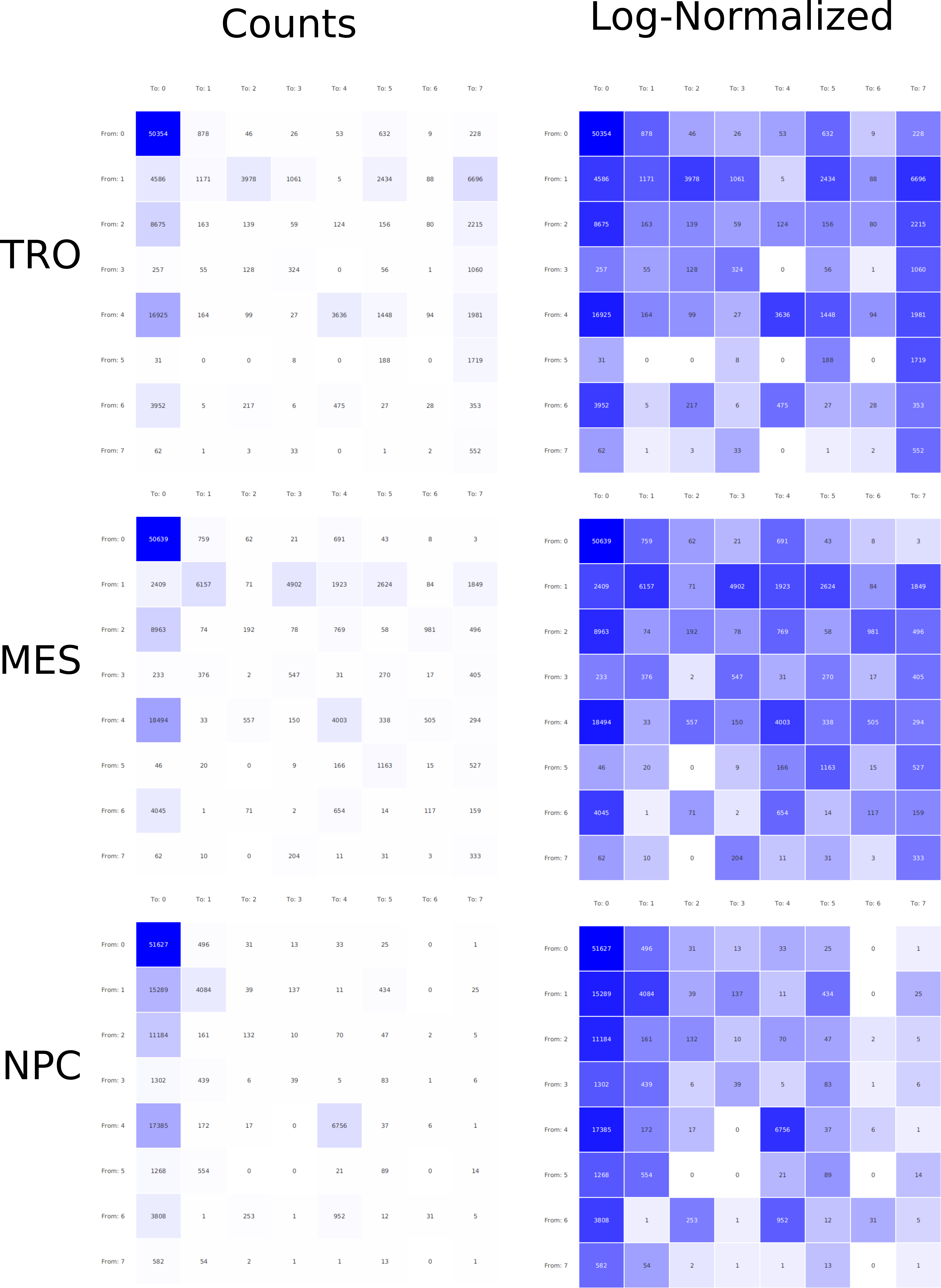
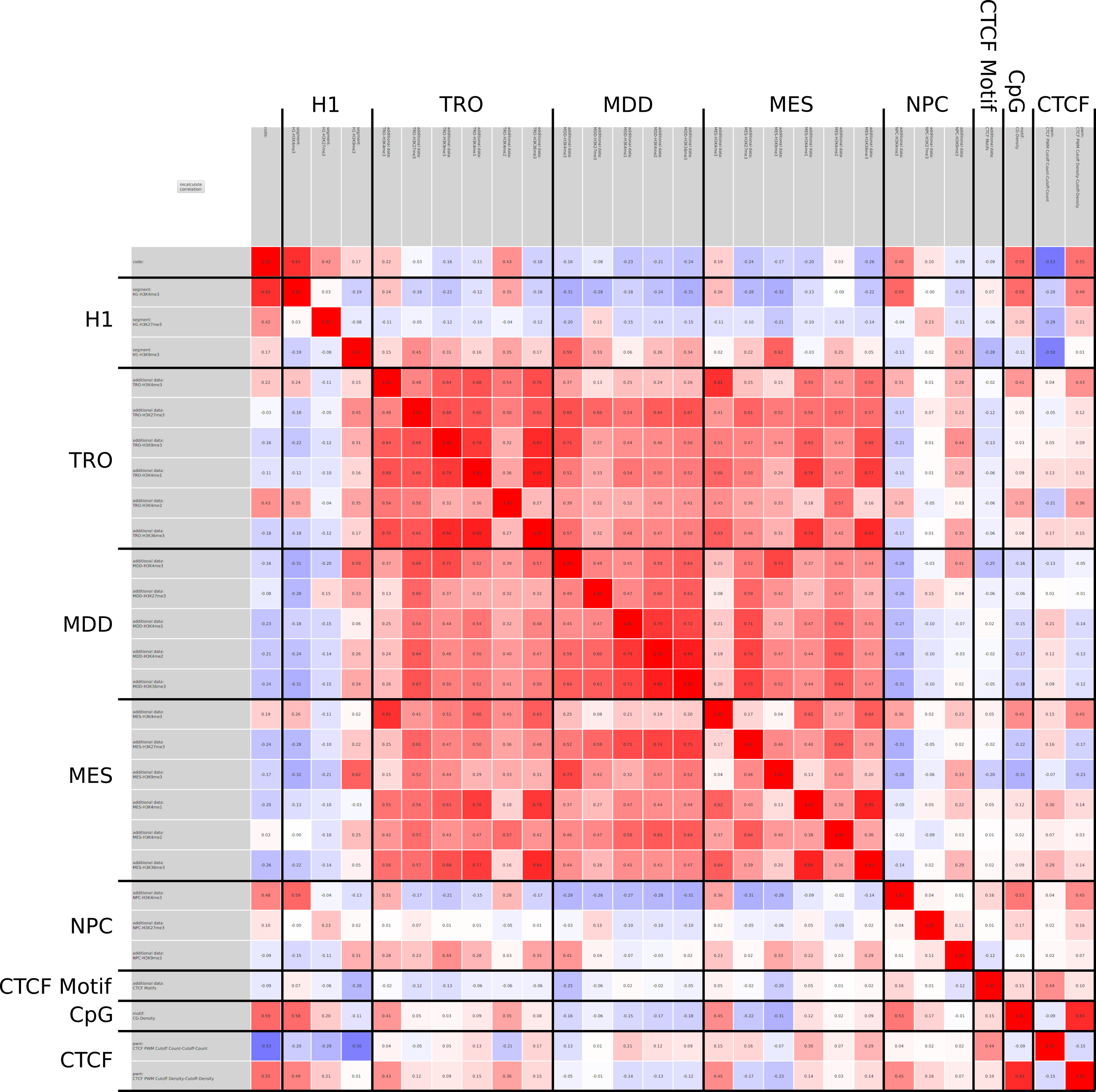
In epigenetics, the change of the combination of histone modifications at the same genomic location during cell differentiation is of great interest for understanding the function of these modifications and their combinations. Besides analyzing them locally for individual genomic locations or globally using correlations between different cells types, intermediate level analyses of these changes are of interest. More specifically, the different distributions of these combinations for different cell types, respectively, are compared to gain new insights.
Results and Discussion
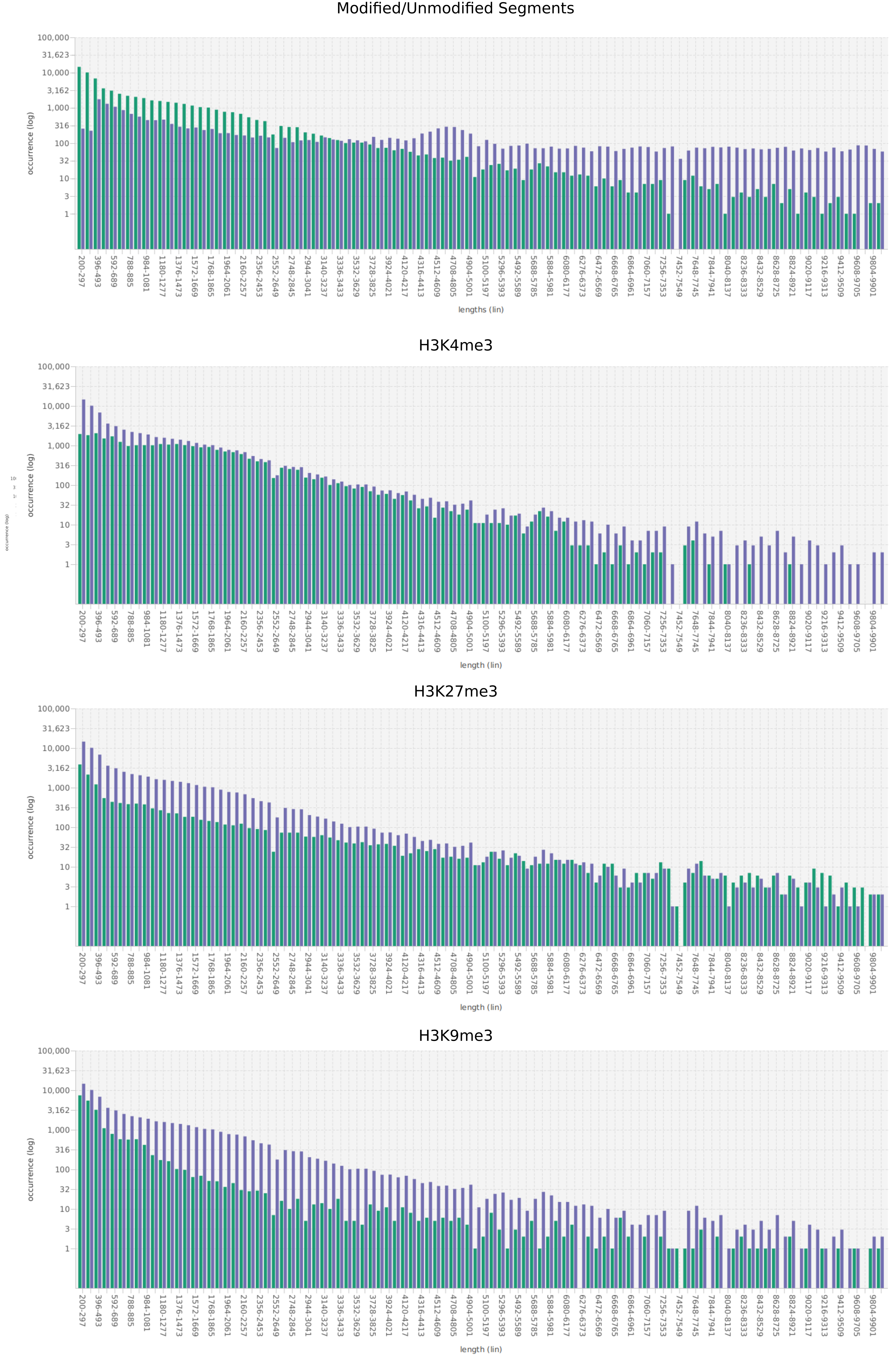
We propose a new tool called Masakari that allows segmenting genomes based on lists of ranges having a certain property, e.g., peaks describing histone modifications. It provides a graphical user interface allowing to select all data sets and setting all parameters needed for the segmentation process. Moreover, the graphical user interface provides statistical graphics allowing to assess the quality and suitability of the segmentation and the selected data.
Conclusion
Masakari provides statistics based visualizations and thus fosters insights into the combination of histone modification marks on genome ranges, and the differences of the distribution of these combinations between different cell types.


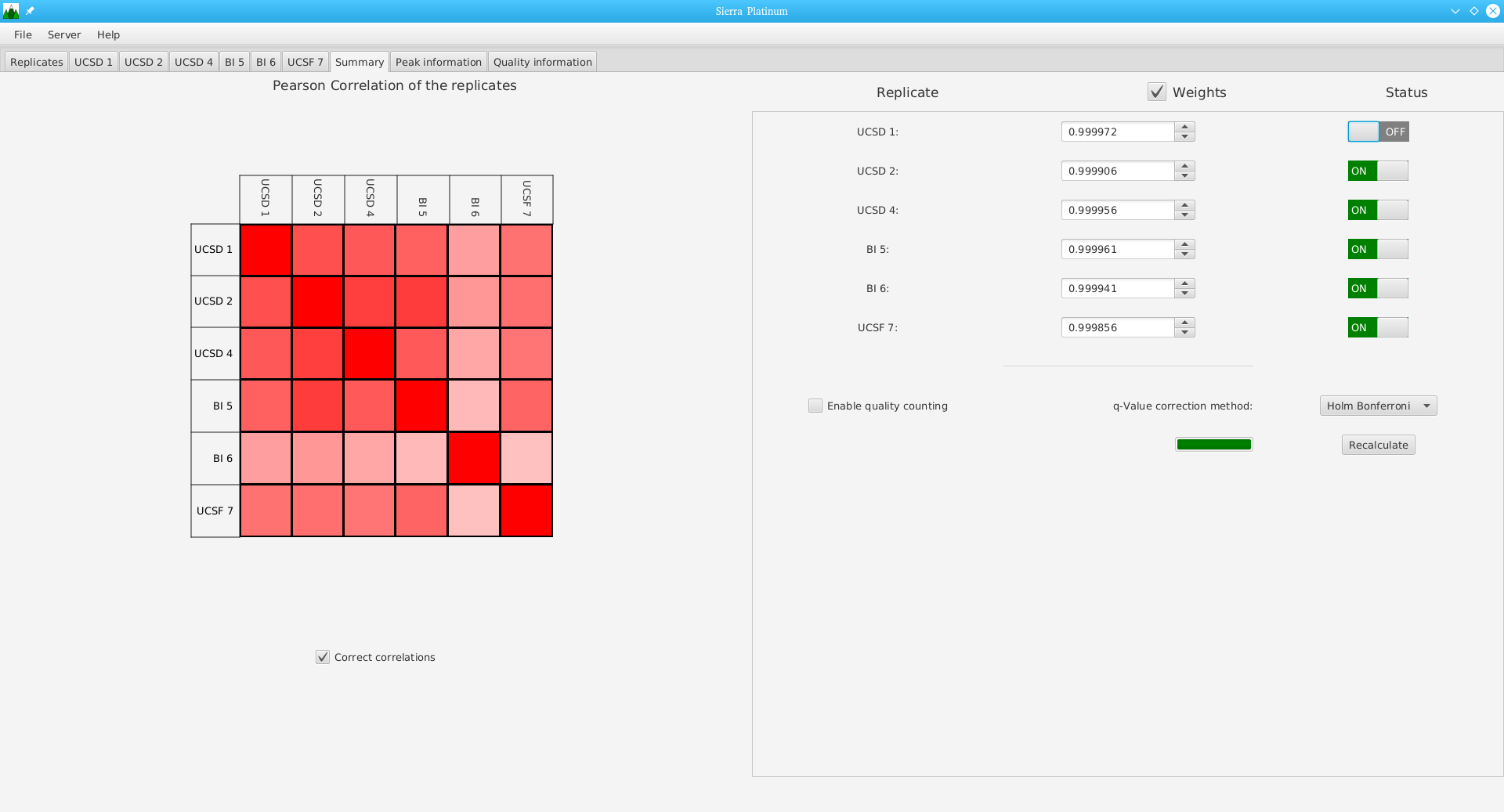
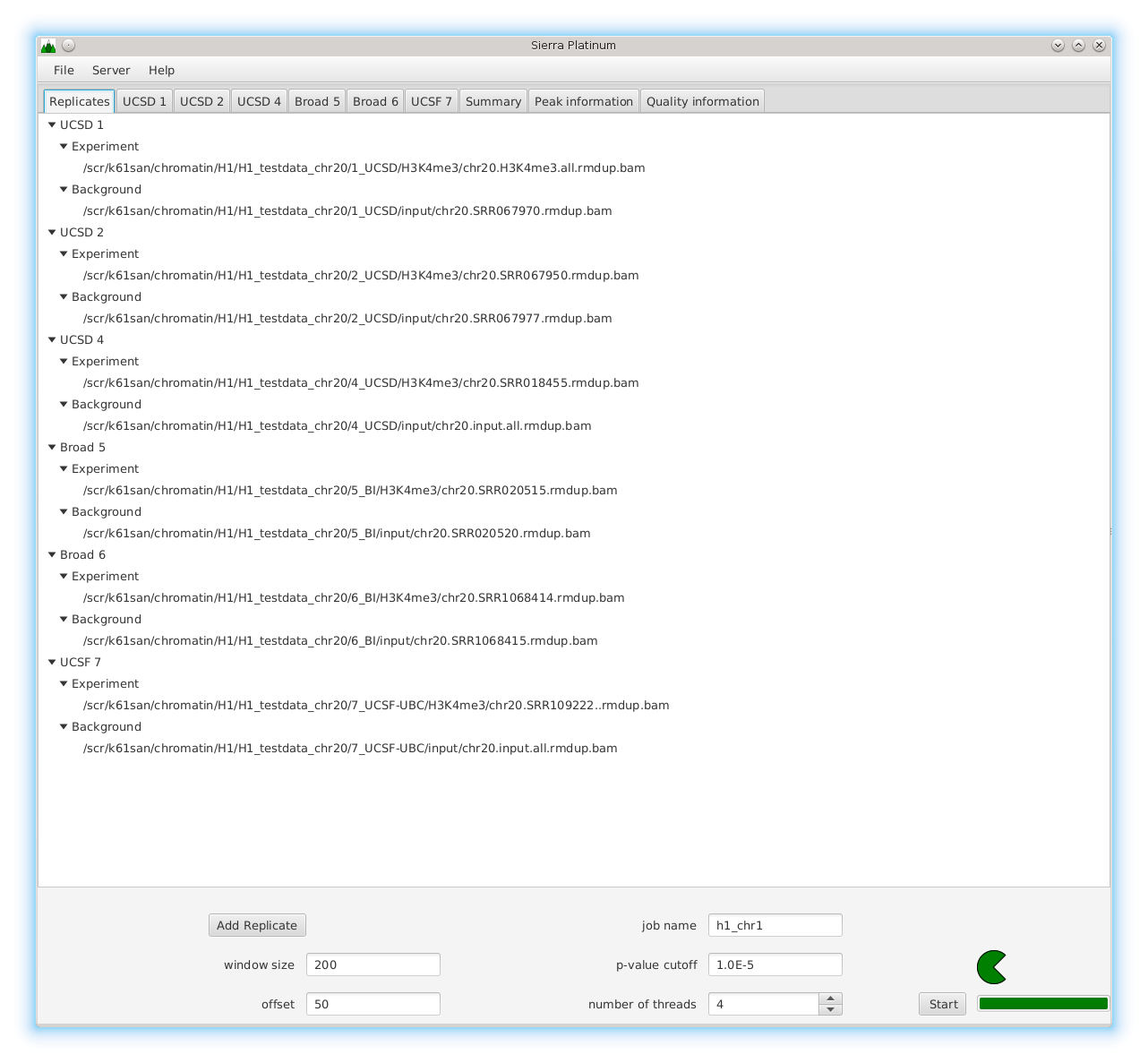
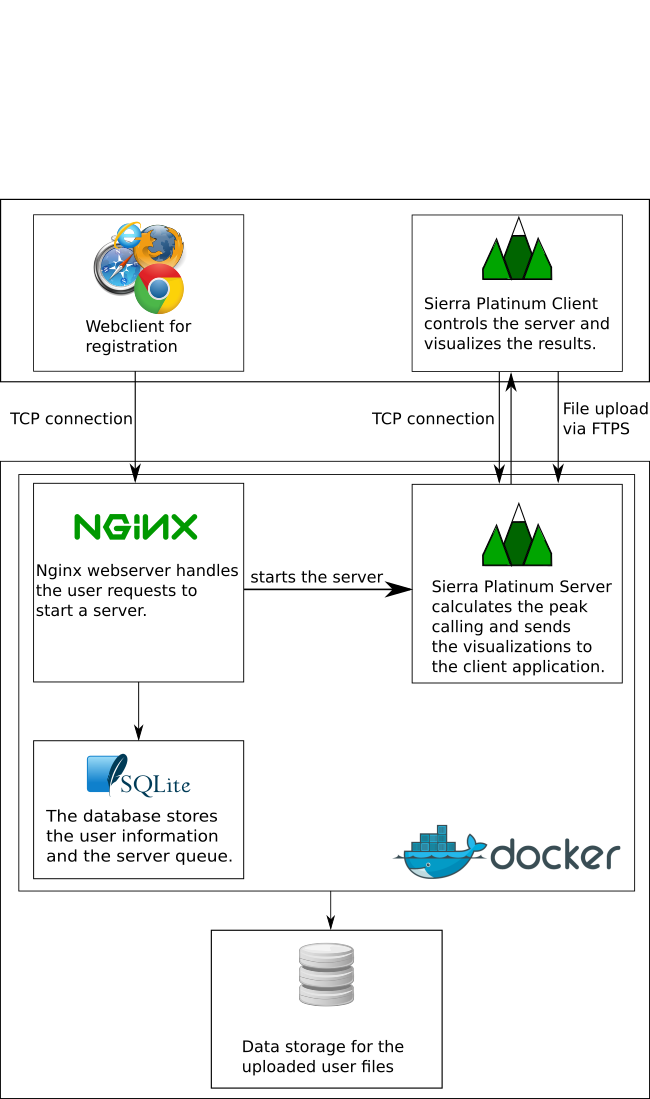
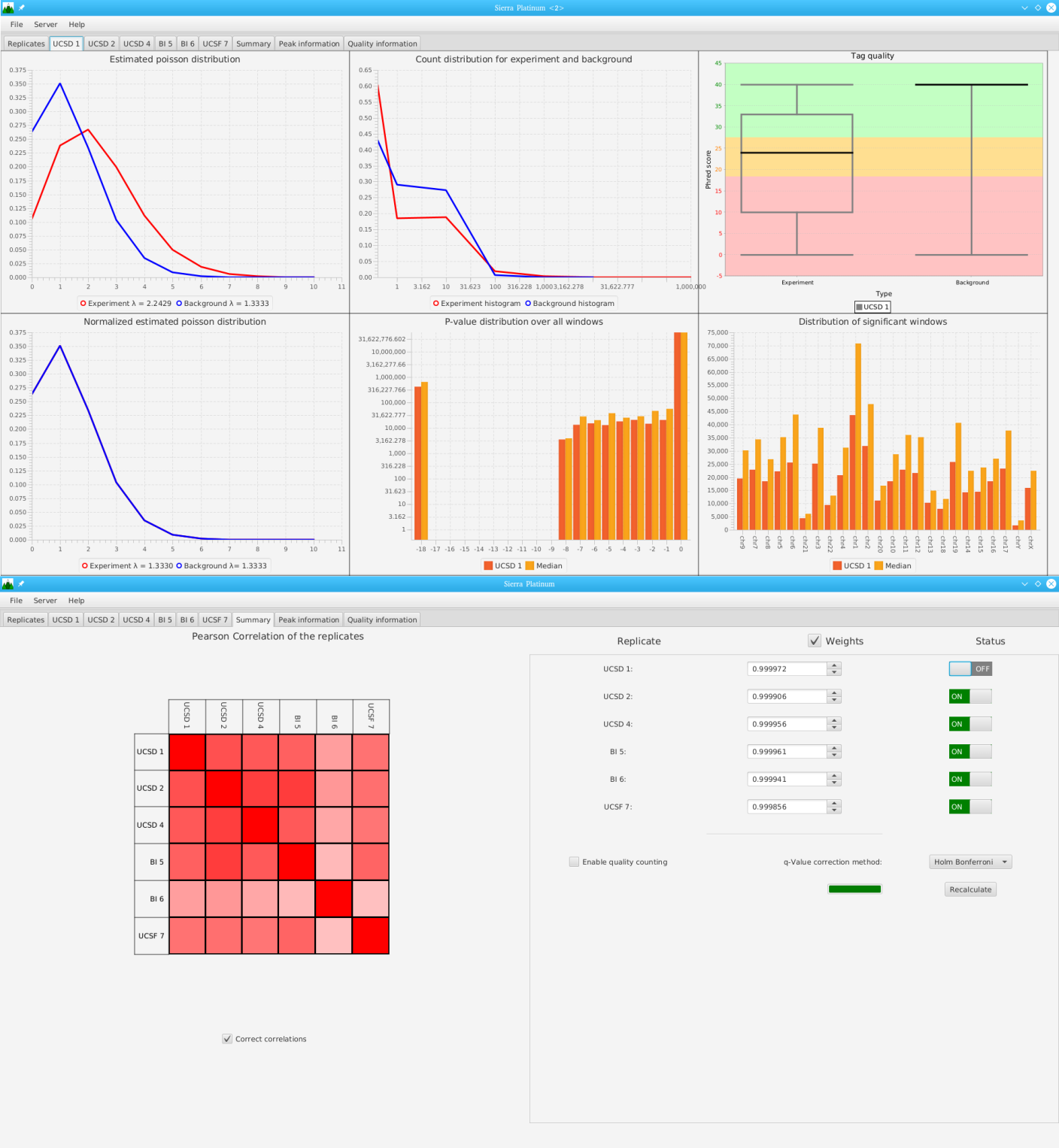
Sierra Platinum is a fast and robust peak-caller for replicated ChIP-seq experiments with visual quality-control and -steering. The required computing resources are optimized but still may exceed the resources available to researchers at biological research institutes. Sierra Platinum Service provides the full functionality of Sierra Platinum: using a web interface, a new instance of the service can be generated. Then experimental data is uploaded and the computation of the peaks is started. Upon completion, the results can be inspected interactively and then downloaded for further analysis, at which point the service terminates.
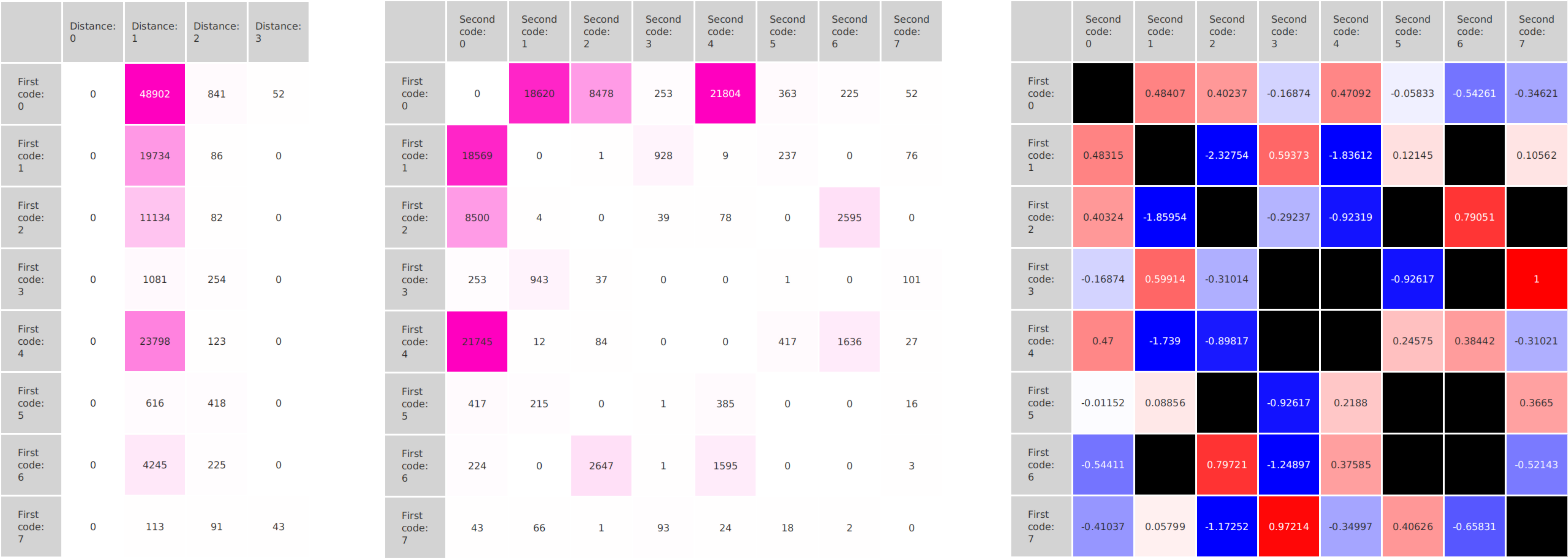
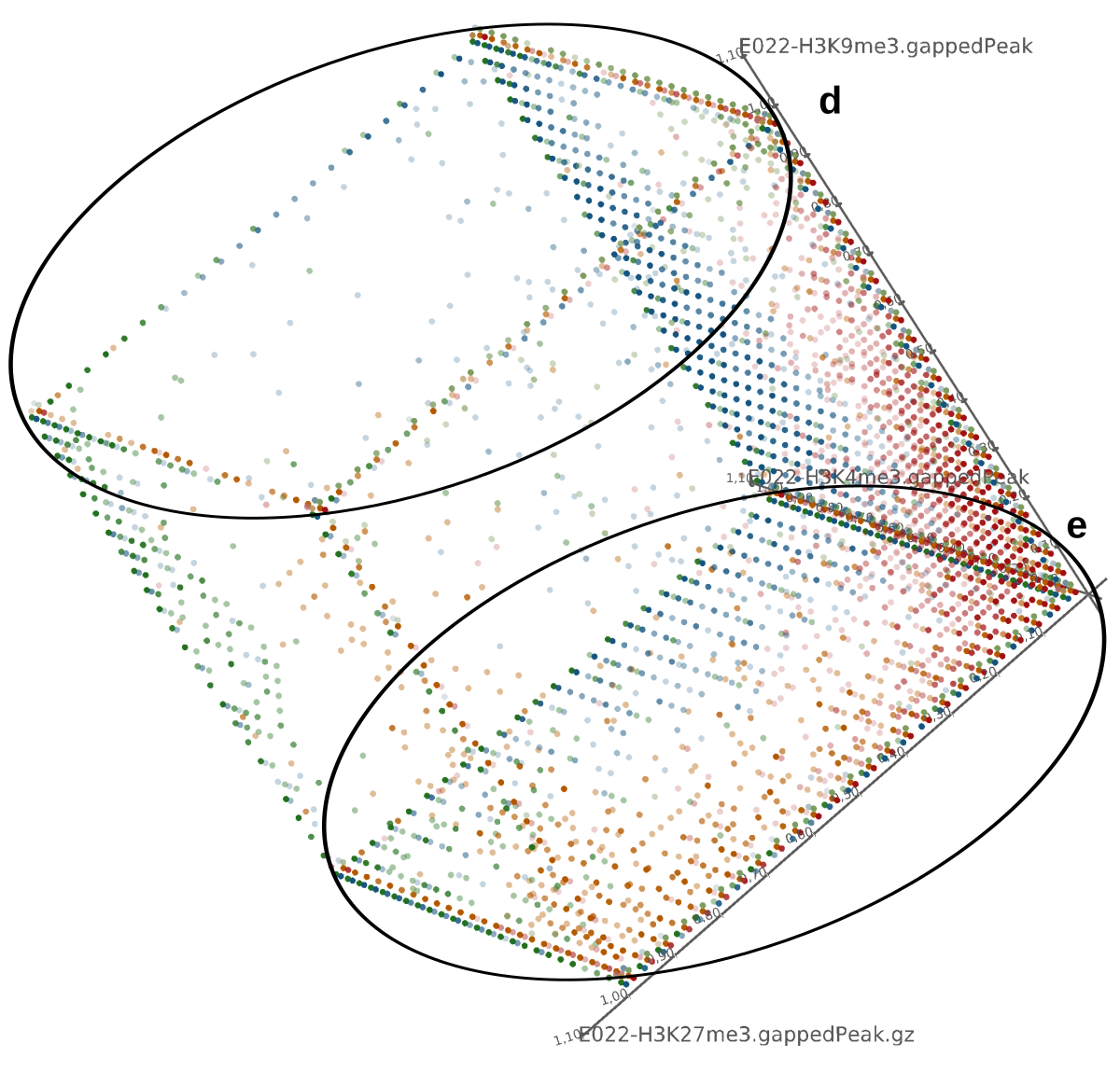
A major goal in epigenetics is understanding how cells differentiate into different cell types. Besides the increase of individual data sets, the amount of replicated experiments generating a tremendous amount of data is ever increasing. While biologists primarily analyze their data on the highest level using statistical correlations or on the lowest level analyzing nucleotide sequences, determining the fate of histone modifications during cell specification necessitates improved analysis capabilities on one or more intermediate levels. For this type of analysis, it proved to be very useful to use tiled binned scatter plot matrices showing binary relationships or to use tiled binned 3D scatter plots showing ternary relationships. Quarternary or general n-ary relationships are not easily analyzable using visualization techniques like scatter plots, only. Therefore, we augmented existing clustering methods with the tiling and binning idea enabling the analysis of n-ary relationships. Analyzing the changes of histone modifications comparing two cell lines using tiled binned clustering, we found new, unknown relations in the data.


Epigenetics data is very important for understand- ing the differentiation of cells into different cell types. More- over, the amount of epigenetic data available was and still is considerably increasing. To cope with this big amount of data, statistical or visual analysis is used. Usually, biologists analyze epigenetic data using statistical methods like correlations on a high level. However, this does not allow to analyze the fate of histone modifications in detail during cell specification or to compare histone modifications in different cell lines. Tiled binned scatter plot matrices proved to be very useful for this type of analysis showing binary relationships. We adapted the idea of tiling and binning scatter plots from 2D to 3D, such that ternary relationships can be depicted. Comparing tiled binned 3D scatter plots—the new method—to tiled binned 2D scatter plot matrices showed, that many relations that are difficult or impossible to find using tiled binned 2D scatter plot matrices can easily be observed using the new approach. We found that using our approach, changes in the distribution of the marks over time (different cell types) or differences between different replicates of the same cell sample are easy to detect. Tiled binned 3D scatter plots proved superior compared to the previously used method due to the reduced amount of overplotting leading to less interaction necessary for gaining similar insights.

DNA bound proteins such as transcription factors and modified histone proteins play an important role in gene regulation. Therefore, their genomic locations are of great interest. Usually, the location is measured using ChIP-seq and analyzed using a peak-caller. While replicated ChIP-seq experiments become more and more available, they are still mostly analyzed using methods based on peak-callers for single replicates. The only exception is PePr, which allows peak calling of several replicates. However, PePr does not provide quality measures to assess the result of the peak-calling process. Moreover, its underlying model might not be suitable for the conditions under which the experiments are performed. We propose a new peak-caller called `Sierra Platinum' that not only allows to call peaks for several replicates but also provides a variety of quality measures. Together with integrated visualizations, the quality measures support the assessment of the replicates and the resulting peaks. We show that Sierra Platinum outperforms methods based on single-replicate peak-callers as well as PePr using a newly generated benchmark data set and using real data from the NIH Roadmap Epigenomics Project.